Genome Annotation is the process to identify, analyze and interpret the relevant information related to the raw DNA sequences. Genome Annotation helps to extract structural location and biological functions of genes. Haemophilus influenza was the first free-living bacterium to be decoded in 1995 by Dr. Owen White. The assembling of the quality reads with a reference genome or de novo assembly in order to obtain the complete genome is a primary requirement for Genome Annotation.
Key: COGs = Clusters of Orthologous Groups, InterPro = Protein Sequence Analysis & Classification, KEGG = Kyoto Encyclopedia of Genes and Genomes, PROSITE = Database of protein domains, families and functional sites, RAST = Rapid Annotation using Subsystems Technology, SEED = The database and infrastructure for comparative genomics, SMART = Simple Modular Architecture Research Tool, UniProt = Universal Protein Resource.
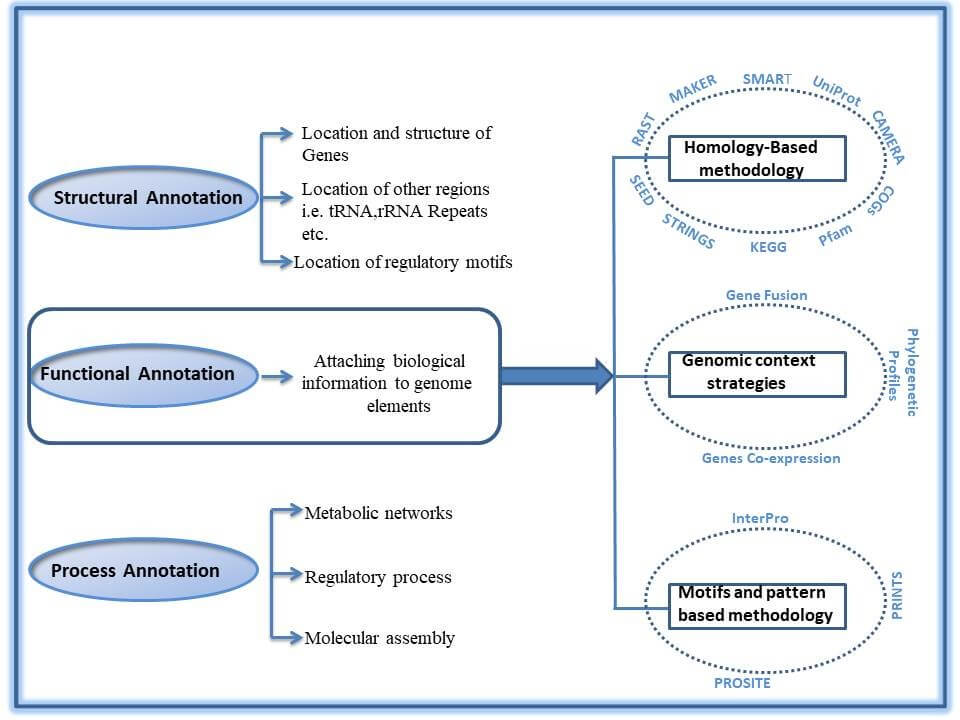
Figure 1 represents that genome annotation is broadly categorized into Structural, Functional and Process Annotation. Structural annotation involves prediction of genes, regulatory motifs, signature elements and others regions i.e. repeats, SNPs, splice sites, non-coding RNA’s, introns etc. Structural annotation can be made with computational techniques using algorithms such as Ab initio-based, Neural Network, Hidden Markov Model and Discriminant.
When structural annotation is completed, the next goal is to understand the function of identified elements of the genome. Functional annotation is the methodology which gives an insight to the function (biological roles) of the signature elements of DNA sequence. Functional annotation is categorized into three strategies i.e. Homology-Based, Genomic Context and Motifs & Patterns Based
Homology-Based method works on the idea that sequence similarities detected between genes or proteins explain that they are homologs i.e. they come from the same ancestor and share the same biochemical function. There are number of databases that provide biological information pertaining to genes and protein, useful to annotate genome. For example, CAMERA’s database (The Community Cyberinfrastructure for Advanced Marine Microbial Ecology Research and Analysis) includes environmental metagenomics and genomic sequence data and software to analyze the environmental samples. There are many more databases (Figure 1) which decode the biological functions based on homology based strategy.
Genomic Context is another important strategy for the functional annotation. In this technique, evolutionary conservation is one of the important properties as we look for the phyletic profiles of protein and gene families. Expression patterns and gene adjacency in genomes are also analyzed by using Genomic Context.
Motifs and patterns based functional annotation strategy, uses the multiple sequence alignment algorithms to identify motifs and patterns shared by multiple input sequences. InterPro, PRINTS and PROSITE are the widely used tools to annotate genomes using motifs and patternsstrategy.
In Process annotation the ultimate goal is to establish a relation among identified productive elements by reconstructing a metabolic networks, acknowledging a regulation process and study at molecular level.








